



This article is part of the AfterMath Data Organizer Electrochemistry Guide
Detailed Description
Like most of the other electrochemical techniques offered by the AfterMath software, Square Wave Voltammetry (SWV) begins with an induction period. During the induction period, a set of initial conditions is applied to the electrochemical cell and the cell is allowed to equilibrate to these conditions. The default initial condition involves holding the working electrode potential at the Initial Potential for a brief period of time (i.e., 3 seconds).
After the induction period, the potential of the working electrode is stepped through a series of forward and reverse pulses from the Initial potential to the Final potential. The forward step is determined by the Square amplitude and the reverse step is determined by subtracting the Square increment from the Square amplitude. Cyclic Square Wave Voltammetry (CSWV) is a variant where the potential of the working electrode is cycled between an Upper potential and a Lower potential.
After the pulse sequence has finished, the experiment concludes with a relaxation period. The default condition during the relaxation period involves holding the working electrode potential at the final potential for an additional brief period of time (i.e., 1 seconds).
At the end of the relaxation period, the post experiment idle conditions are applied to the cell and the instrument returns to the idle state.
Difference current between the forward and reverse pulses is plotted as a function of the potential applied to the working electrode, resulting in a voltammogram.
Parameter Setup
The parameters for this method are arranged on various tabs on the setup panel. The most commonly used parameters are on the Basic tab, and less commonly used parameters are on the Advanced tab. Additional tabs for Ranges and Post experiment idle conditions are common to all of the electrochemical techniques supported by the AfterMath software.
Basic Tab
For SWV, you can click on the “I Feel Lucky” button (located at the top of the setup) to fill in all the parameters with typical default values (see Figure 1). You may need to change the Initial potential and Final potential, to values which are appropriate for the electrochemical system being studied.

Figure 1 : Basic setup for SWV.
The waveform that is applied to the electrode is a series of forward and reverse pulses (see Figure 2) each having an amplitude of Square amplitude and incremented according to the Square increment. The total time for the forward and reverse pulses is the Square period. Using the sample waveform below, the first pulse is 





Figure 2: Zoom of waveform for SWV.
Advanced Tab
The Advanced Tab for this method allows you to change the behavior of the potentiostat during the induction period and relaxation period. By default, the potential applied to the working electrode during the induction and relaxation period will match the initial potential and final potential, respectively, as specified on the Basic Tab. You may override this default behavior, and you may also change the durations of the induction and relaxation periods if you wish.
Ranges Tab
Though AfterMath has the ability to automatically select the appropriate ranges for voltage and current during an experiment it is best to manually select the current range for any pulse technique. Please see the separate discussions on autoranging and the Ranges Tab for more information.
Post Experiment Conditions Tab
After the Relaxation Period, the Post Experiment Conditions are applied to the cell. Typically, the cell is disconnected but you may also specify the conditions applied to the cell. Please see the separate discussion on post experiment conditions for more information.
Typical Results
Typical results for a 





Figure 3:Square Wave Voltammogram of a Ferrocene Solution


Figure 4: A) Forward current and B) Reverse current of a Ferrocene Solution
Below is an example for CSWV of a

Figure 5 : Cyclic Square Wave Voltammogram for a Ferrocene Solution
Theory
The following is a brief introduction to the theory of SWV. SWV was invented by Ramaley and Krause1. Please see Bard and Faulkner2, Osteryoung and O'Dea 3 or Osteryoung and Osteryoung4 for additional information on the technique. CSWV, originally developed by Xinsheng and Guogang5, and recently revived by Helfrick and Bottomley6, is covered in the literature also.
SWV combines the aspects of several pulse voltammetric methods, including the background supression and sensitivity of DPV, the diagnostic value of NPV, and the ability to interrogate products directly in the manner of RNPV.
Consider a reaction 

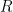

The reverse pulse consists of stepping the potential of the working electrode to more positive values and the current is sampled near the end of the reverse pulse. The difference current is calculated by subtracting the reverse current from the forward current.
As the potential of the working electrode approaches
SWV's strengths lie in diagnostics, meaning that it is not typically used for quantitation. It is possible however, to calculate peak height using the equation

where 


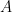







Table 1. Dimensionless Peak Current (
 | ||||
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
| |
 |
 |
 |
 |
| |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
 |
aData from reference 3.
 = Square amplitude
= Square amplitude
 corresponds to SCV
corresponds to SCV
 = Square increment
= Square increment
Applications
The first application uses SWV to monitor binding events associated with electrochemical sensors. White and Plaxco7 developed redox-tagged electrochemical sensors from electrode-bound oligonucleotides. Tuning the frequency of the voltammetric measurements allows the researchers to amplify both the unbound and target-bound signals, essentially, making an “On/Off” sensor.
The second example used several electrochemical techniques, SWV among them, to monitor metal dissociation events. Chakrabarti et al.8 combined RDE with anodic stripping voltammetry to distinguish between labile and nonlabile complexes in extremely low concentrations in aqueous solutions and in precipitation samples. The authors then showed that DPV, SCV and SWV all give similar dissociation constants but SWV's sensitivity was two orders of magnitude higher than SCV. This is a nice example of how SWV compares with other electrochemical techniques.
References
1. Ramaley, L.; Krause, Jr., M. S. Anal. Chem. 1969, 41, 1365–1369.
2. Faulkner, L. R.; Bard, A. J. Polarography and Pulse Voltammetry, Electrochemical Methods: Fundamentals and Applications, 2nd ed.; Wiley: New Jersey, 2000; 261-304.
3. Osteryoung, J.; O'Dea, J. J. Electroanal. Chem. 1986, 14, 209.
4. Osteryoung, J.; Osteryoung, R. A. Anal. Chem. 1985, 57, 101A–110A.
5. Xinsheng, C.; Guogang, P. Anal. Lett. 1987, 20, 1511-1519.
6. Helfrick, Jr., J. C.; Bottomley, L. A. Anal. Chem. 2009, 81, 9041-9047.
Additional Resources
Read More
Post-Experiment Idle Conditions
This article complements the AfterMath Data Organizer Electrochemistry Guide
When an experiment concludes, the instrument reverts to its idle mode and applies a set of idle conditions to the electrochemical cell.
By default, the instrument simply disconnects from the electrochemical cell.
You may override this default behavior by specifying an alternate set of conditions on the Post-Experiment tab on the form shown below. In the example below, the first working electrode is placed under galvanostatic control after the experiment concludes, and a signal level (


Links: Electrochemist's Guide, AfterMath User's Guide, AfterMath Main Support Page
Related Topics: cell switching, relaxation period, idle conditions
Range Tab
This article is part of the AfterMath Data Organizer Electrochemistry Guide
Autoranging
The choice of measurement sensitivity for the potential and current signals is made on the Ranges Tab. By default, both these range settings are set to “Auto”, meaning that the software and/or instrument will attempt to choose an appropriate signal sensitivity automatically. This is referred to as the “autorange” feature.

Figure 1: Choosing Specific Ranges
In the example shown above (see Figure 1), the potential and current ranges on the second working electrode are set to autorange.
The potential range on the first working electrode has been manually set to 


Also in the example above, the user is in the process of selecting a current range for the first working electrode from a drop-down menu. The four choices shown in the menu (

Overriding Autorange
In some cases, you will need to override the “Auto” range feature and select a particular range that is known to be appropriate for the particular electrochemical system you are studying. Consider a 







Figure 2: Influence of Current Range Choice on Voltammogram Quality
Not all instruments support the “autorange” feature. For instruments which do not support this feature, choosing the “Auto” range setting usually causes the least sensitive range setting to be selected.
Additional Resources
Links: Electrochemist's Guide, AfterMath User's Guide, AfterMath Main Support Page
Related Topics: autoranging
Ranges
This article complements the AfterMath Data Organizer Electrochemistry Guide
The “Ranges” tab is set to Autorange by default (see below).
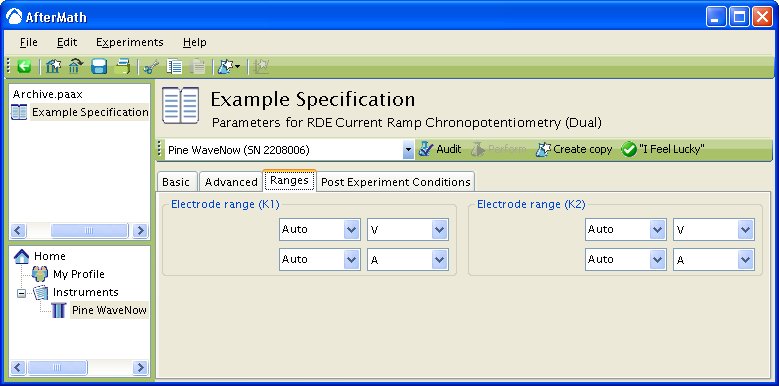
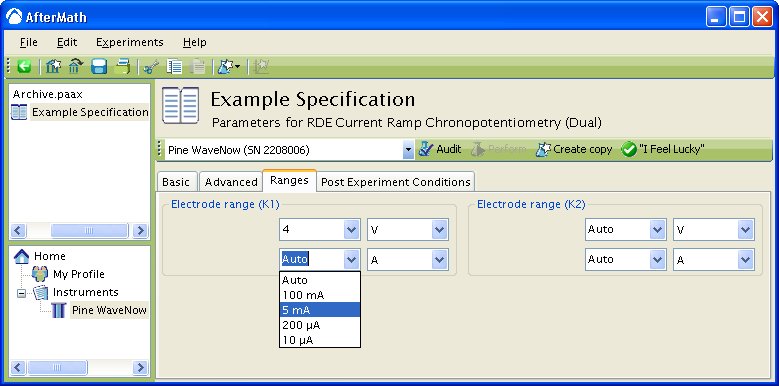
To change the default settings, simply choose the wanted potential or current range from the drop down menu. In the example below, 
Normal Pulse Voltammetry (NPV)
This article is part of the AfterMath Data Organizer Electrochemistry Guide
Detailed Description
Like most of the other electrochemical techniques offered by the AfterMath software, this experiment begins with an induction period. During the induction period, a set of initial conditions is applied to the electrochemical cell and the cell is allowed to equilibrate to these conditions. The default initial condition involves holding the working electrode potential at the Baseline Potential for a brief period of time (i.e., 3 seconds).
After the induction period, the potential of the working electrode is stepped through a series of pulses from the Initial potential to the Final potential. The potential is incremented with each successive pulse according to the Pulse increment. Current is measured at the time obtained by subtracting the Sample window from the Pulse width. Cyclic Normal Pulse Voltammetry (CNPV) consists of a cycling (through a series of potential pulses also) the potential of the working electrode between an Upper potential and a Lower potential. Reverse Normal Pulse Voltammetry (RNPV) is a variant of NPV where you choose to apply a Baseline potential in a region where faradaic current is flowing at a maximal rate (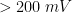
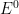
After the pulse sequence has finished, the experiment concludes with a relaxation period. The default condition during the relaxation period involves holding the working electrode potential at the Final potential for an additional brief period of time (i.e., 1 seconds).
At the end of the relaxation period, the post experiment idle conditions are applied to the cell and the instrument returns to the idle state.
Current is sampled during each pulse and is plotted as a function of the potential applied to the working electrode, resulting in a voltammogram.
Parameter Setup
The parameters for this method are arranged on various tabs on the setup panel. The most commonly used parameters are on the Basic tab, and less commonly used parameters are on the Advanced tab. Additional tabs for Ranges and Post experiment idle conditions are common to all of the electrochemical techniques supported by the AfterMath software.
Basic Tab
For NPV, you can click on the “I Feel Lucky” button (located at the top of the setup) to fill in all the parameters with typical default values (see Figure 1). You may need to change the Baseline potential, Initial potential and Final potential, to values which are appropriate for the electrochemical system being studied.

Figure 1: Basic setup for NPV.
There is no separate Experiment to choose for RNPV, rather you are entering the parameters so as to begin in a region where a faradaic current flows at a maximal rate (

Figure 2: Basic setup for RNPV.
For CNPV, you can click on the “I Feel Lucky” button (located at the top of the setup) to fill in all the parameters with typical default values (see Figure 3). You may need to change the Baseline potential, Number of segments, Initial potential, Upper potential, Lower potential, and Final potential, to values which are appropriate for the electrochemical system being studied.
If you choose any odd number of segments greater than two, the parameters that must be entered are a little different than the two segment case. You must choose a Baseline potential, Initial Potential, Upper Potential, Lower Potential, and Final potential. You must also choose whether the Initial direction is rising (pulses toward Upper Potential) or falling (pulses toward Lower Potential). If the Initial direction is rising, the Final potential must be different than the Lower potential. If the Initial direction is falling, the Initial potential must be different than the Lower potential.
If you choose any even number of segments greater than two, the parameters that must be entered are the same as the three segment case. You must choose an Baseline potential, Initial Potential, Upper Potential, Lower Potential, and Final potential. You must also choose whether the Initial direction is rising (pulses toward Upper Potential) or falling (pulses toward Lower Potential). If the Initial direction is rising, the Final potential must be different than the Upper potential. If the Initial direction is falling, the Initial potential must be different than the Lower potential.

Figure 3: Basic setup for CNPV.
The waveform that is applied to the electrode for all three techniques consists of a series of Pulse periods with a potential step, for a specified time, from a baseline potential near the end of each period (see Figure 4). The potential step is incremented with each period until the final potential is reached. During each potential pulse, the current is measured at a specified time before the end of the pulse. For CNPV, the pulse sequence starts at the Initial potential and is then cycled between the Upper potential and Lower potential for one less than the specified number of segments. The final segment steps from the Upper potential or Lower potential to the Final potential depending if the Initial direction was rising or falling.


Figure 4 : Waveform. Orange trace – applied potential, red squares – current sampled. A: Waveform. B: Zoom of Waveform.
Advanced Tab
The Advanced Tab for this method allows you to change the behavior of the potentiostat during the induction period and relaxation period. By default, the potential applied to the working electrode during the induction and relaxation period will match the initial potential and final potential, respectively, as specified on the Basic Tab. You may override this default behavior, and you may also change the durations of the induction and relaxation periods if you wish.
Ranges Tab
Though AfterMath has the ability to automatically select the appropriate ranges for voltage and current during an experiment it is best to manually select the current range for any pulse technique. Please see the separate discussions on autoranging and the Ranges Tab for more information.
Post Experiment Conditions Tab
After the Relaxation Period, the Post Experiment Conditions are applied to the cell. Typically, the cell is disconnected but you may also specify the conditions applied to the cell. Please see the separate discussion on post experiment conditions for more information.
Typical Results
The typical results for NPV of a 








Figure 5: Typical NPV results for
The typical results for RNPV of the same 


Figure 6: Typical RNPV results for a Ferrocene Solution.
Finally, the typical results for CNPV (two segments) of the same

Figure 7: Typical CNPV results for a Ferrocene Solution.
Theory
The following is a brief introduction to the theory of NPV. Please see Bard and Faulkner1 for additional information on the technique. NPV is a derivative technique of Normal Pulse Polarography (NPP). NPP is a technique that was traditionally used with Dropping Mercury Electrodes and Static Mercury Dropping Electrodes. The waveform for the two techniques is the same, however it is appropriate to use the term “Normal Pulse Voltammetry” when referring to the application of the waveform to nonpolarographic electrodes.
Consider a reaction 


The potential pulse is incrementally increased with each cycle. As the potential of the working electrode approaches

where 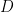

As seen in the Typical Results section, RNPV gives the same wave shape but not the same current. These results are analogous to DPSCA where the current during the forward pulse is different than the current in the reverse pulse. Here in RNPV, the baseline potential is such that
![i_{d,RPV} = \frac{nFAD_O^{1/2}C_O^*}{{\pi}^{1/2}}\left[{\frac{1}{({\tau}-{\tau}')^{1/2}}}-{\frac{1}{{\tau}^{1/2}}}\right]](https://s0.wp.com/latex.php?latex=i_%7Bd%2CRPV%7D+%3D+%5Cfrac%7BnFAD_O%5E%7B1%2F2%7DC_O%5E%2A%7D%7B%7B%5Cpi%7D%5E%7B1%2F2%7D%7D%5Cleft%5B%7B%5Cfrac%7B1%7D%7B%28%7B%5Ctau%7D-%7B%5Ctau%7D%27%29%5E%7B1%2F2%7D%7D%7D-%7B%5Cfrac%7B1%7D%7B%7B%5Ctau%7D%5E%7B1%2F2%7D%7D%7D%5Cright%5D&bg=ffffff&fg=000&s=3&c=20201002)
where the parameters are as described above. Notice that in the Typical Results section, there is a slight anodic current flowing at the beginning of the experiment. The magnitude of this current is given by the equation

where the parameters are as described above. The magnitude of this current is the difference between the NPV and RNPV currents.
Application
The first example uses NPV to confirm a diffusion coefficient calculated initially by CA. Oyaizu et al.2 produced an organic radical polymer to be used in charge-storage applications. The current in this case is controlled by diffusion of the counter ion through the film.
The second example also uses NPV to measure a diffusion coefficient. Welch et al.3 measured the diffusion coefficients of a free and DNA-bound organo-metallic complex. NPV was superior to CV in this instance due to the low concentration of species in solution.
The next example uses RNPV. RNPV's useless lies in its ability to examine products from chemical reactions that take place after an electrochemical reaction. Osteryoung et al.4 used RNPV to obtain the backwards rate constant and equilibrium constant for the dimerization of N-methyl-2-carbomethoxypyridinium radical, produced after the electrochemical reduction of the N-methyl-2-carbomethoxypyridinium ion.
References
1. Faulkner, L. R.; Bard, A. J. Polarography and Pulse Voltammetry, Electrochemical Methods: Fundamentals and Applications, 2nd ed.; Wiley: New Jersey, 2000; 261-304.
2. Oyaizu, K.; Ando, Y.; Konishi, H.; Nishide, H. J. Am. Chem. Soc. 2008, 130, 14459–14461.
3. Welch, T. W.; Corbett, A. H.; Thorp, H. H. J. Phys. Chem. 1995, 99, 11757–11763.
4. Osteryoung, J.; Talmor, D.; Hermolin, J.; Kirowa-Eisner, E. J. Phys. Chem. 1981, 85, 285–289.
Additional Resources
Links: Electrochemist's Guide, AfterMath User's Guide, AfterMath Main Support Page
Related Techniques: Differential Pulse Voltammetry (DPV), Square Wave Voltammetry (SWV)
Read More
Open Circuit Potential (OCP)
This article is part of the AfterMath Data Organizer Electrochemistry Guide
Detailed Description
Like most other electrochemical techniques, this experiment begins with an induction period. During the induction period, a set of initial conditions which you specify is applied to the electrochemical cell and the cell is allowed to equilibrate to these conditions. Data are not collected during the induction period.
After the induction period, the potential difference between the working and counter electrodes is monitored for a specified period of time.
The experiment concludes with a relaxation period. During the relaxation period, a set of final conditions which you specify is applied to the electrochemical cell and the cell is allowed to equilibrate to these conditions. Data are not collected during the relaxation period.
At the end of the relaxation period, the post-experiment conditions are applied to the cell, and the instrument returns to the idle state.
Potential is plotted as a function of time.
Parameter Setup
The parameters for this method are arranged on two tabs on the setup panel. The Basic tab contains the parameters relating to the electrolysis. An additional tab for Post Experiment Conditions is common to all of the electrochemical techniques supported by the AfterMath software.
Basic Parameters Tab
You can click on the “I Feel Lucky” button (located at the top of the setup) to fill in all the parameters with typical default values (see Figure 1). You may want to change the Duration in the Electrolysis period box to a value which is appropriate for the electrochemical system being studied. You may also want to change the Number of intervals in the Sampling Control box.

Figure 1: Basic Setup for OCP.
Post Experiment Conditions Tab
After the Relaxation Period, the Post Experiment Conditions are applied to the cell. Typically, the cell is disconnected but you may also specify the conditions applied to the cell. Please see the separate discussion on post experiment conditions for more information.
Typical Results
Figure 2 shows the typical results for a solution of 

Figure 2: Open Circuit Potentiogram of a Potassium Ferricyanide Solution
Theory
Consider the reaction 
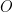
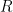

![E = E^0- {\frac{RT}{nF}} \; ln \left({\frac{[R]}{[O]}}\right)](https://s0.wp.com/latex.php?latex=E+%3D+E%5E0-+%7B%5Cfrac%7BRT%7D%7BnF%7D%7D+%5C%3B+ln+%5Cleft%28%7B%5Cfrac%7B%5BR%5D%7D%7B%5BO%5D%7D%7D%5Cright%29&bg=ffffff&fg=000&s=3&c=20201002)
where 


Additional Resources
Links: Electrochemist's Guide, AfterMath User's Guide, AfterMath Main Support Page
Related Techniques: Bulk Electrolysis (BE)
Scope of this Guide

This guide is intended for owners of the AfterMath software package who use the software to control electrochemical instrumentation manufactured by Pine Research Instrumentation.
The reader is assumed to have a good working knowledge of electrochemistry (voltammetry, electrochemical cells, corrosion, etc.).
The reader is also assumed to have a operational knowledge of other non-electrochemical features of the AfterMath software package. These other features of AfterMath are described in the primary AfterMath User's Guide and they are not discussed here. The Electrochemist's Guide to AfterMath deals only with issues pertaining to the use of AfterMath with an electrochemical potentiostat/galvanostat system.
Related Links: Electrochemist's Guide, AfterMath User's Guide, AfterMath Main Support Page
Koutecky-Levich Rotating Disk Electrochemistry (KL-RDE)
This article is part of the AfterMath Data Organizer Electrochemistry Guide
Detailed Description
Like most of the other electrochemical techniques offered by the AfterMath software, this experiment begins with an induction period. During the induction period, a set of initial conditions is applied to the electrochemical cell and the cell is allowed to equilibrate to these conditions. The default initial condition involves holding the working electrode potential at the Initial Potential for a brief period of time (i.e., 3 seconds). Since the potentiostat is being used to control the rotator speed, the rotator is also spun at the desired Initial speed during this time.
At each rotation speed, the potential of the rotating disk is cycled according to the parameters the you entered. After the sweep has finished at the final rotation speed, the experiment concludes with a relaxation period. The default condition during the relaxation period involves holding the working electrode potential at the final potential for an additional brief period of time (i.e., 1 seconds).
At the end of the relaxation period, the post experiment idle conditions are applied to the cell and the instrument returns to the idle state.
Current is plotted as a function of the potential applied to the rotating disk for each rotation speed, resulting in a series of voltammograms.
Parameter Setup
The parameters for this method are arranged on various tabs on the setup panel. The most commonly used parameters are on the Basic tab, and less commonly used parameters are on the Advanced tab. Additional tabs for Ranges and Post Experiment Conditions are common to all of the electrochemical techniques supported by the AfterMath software.
Basic Tab
You can click on the “I Feel Lucky” button (located at the top of the setup) to fill in all the parameters with typical default values. You will no doubt need to change the Initial Potential, Final Potential, and Sweep Rate to values which are appropriate for the electrochemical system being studied.
The Basic tab contains the same parameters as RDE except that the Rotator Parameters box contains an Initial speed, a Final speed, an Increment method, and an Iterations (see Figure 1). The rotation speeds will be chosen based on the Increment method and the number of Iterations. The different Increment methods are Linear, Levich, Koutecky, Eisenberg and Custom (see Figure 2). When Linear is chosen the rotation speeds will be evenly spaced between the Initial speed and Final speed. When Levich is chosen, the increments will be spaced such that the increases in the limiting current (





KL-RDE is typically done in order to obtain a heterogeneous rate constant for the species of interest. Therefore, typically you choose one segment and enter an Initial potential, Final potential, Sweep rate, and Rotator Speed. Please see the webpage regarding RDE if more than one segment is needed.

Figure 1: Basic Setup Tab with Linear Increment Method Chosen.

Figure 2: Basic Setup Tab Showing Different Increment Methods.
The Electrode Range on the Basic tab is used to specify the expected range of current. If the choice of electrode range is too small, actual current may go off scale and be truncated. If the electrode range is too large, the voltammogram may have a noisy, choppy, or quantized appearance. Please see the ugly duckling webpage for more information.
Advanced Tab
The Advanced Tab for this method (see Figure 3) is the same as the Advanced Tab for RDE and allows you to change the behavior of the potentiostat during the induction period and relaxation period. By default, the potential applied to the working electrode during the induction and relaxation period will match the initial potential and final potential, respectively, as specified on the Basic Tab. You may override this default behavior, and you may also change the durations of the induction and relaxation periods if you wish.
Other important parameters on the Advanced tab are found in the Sampling Control area. This area contains two parameters, Alpha and Threshold which control when and how samples are acquired during the sweep portion of the experiment.

Figure 3: Advanced Parameters for KL-RDE
As mentioned previously, the waveform applied to the electrode (see Figure 4A for a two-segment example) is not truly linear. The actual waveform is a staircase of small potential steps (see Figure 4B). The duration of each small step is called the step period, and the step period is automatically chosen to take full advantage of the resolution of the potentiostat's digital-to-analog converter.


Figure 4: Two Segment RDE Waveform Detailing the A) Total waveform and B) Magnified Waveform Showing the Applied Potential Steps (black trace) and Measured Potential (red trace)
The Alpha parameter controls the exact time within the step period at which the current is sampled. A alpha value of zero means the current is sampled at the start of the step period, immediately after a new potential step is applied. An alpha value of 100 means the current is sampled at the end of the step period, immediately before the next potential step is applied.
Changing alpha will have little effect on the voltammogram for a freely diffusing species in solution; however, variations in alpha can dramatically influence the results for surface bound species, especially when using older potentiostats with low DAC resolution (i.e., 12-bit).
Newer potentiostats (such as the WaveNow and WaveNano portable USB potentiostats) have 16-bit DAC resolution, so voltammograms acquired using these instruments are less influenced by the choice of alpha value. Nevertheless, researchers who use digital potentiostats to study surface-confined electrochemical systems (rather than freely diffusing species in solution) should be aware of the influence of this parameter. Further details can be found in the literature1 and in a related article about CBP Bipotentiostat Interface Boards.
The Threshold parameter helps you to limit the amount of data retained as the voltammogram is acquired. The threshold parameter controls the interval between samples as the potential is swept from one limit to another. By default, a data point is acquired every time the sweep moves 5 millivolts. You can change the threshold from 5 millivolts to a smaller interval (if you want to acquire more data) or to a greater interval (if you want to acquire less data).
Extreme values for the threshold parameter can lead to undesirable results (see Figure 7 of cyclic voltammetry). A very small Threshold value will produce smooth curves yet results in large files. A very large value though, results in jagged curves.
Ranges Tab
AfterMath has the ability to automatically select the appropriate ranges for voltage and current during an experiment. However, you can also choose to enter the voltage and current ranges for an experiment. Please see the separate discussions on autoranging and the Ranges Tab for more information.
Post Experiment Conditions Tab
After the Relaxation Period, the Post Experiment Conditions are applied to the cell. Typically, the cell is disconnected but you may also specify the conditions applied to the cell. Please see the separate discussion on post experiment conditions for more information.
Typical Results
Consider a 




Figure 5: KL-RDE Results for a Ferrocene Solution
You can add a peak height tool to each of the voltammograms in order to obtain limiting currents (ilim) at each rotation speed. The example below, taken from the RDE webpage, illustrates how to determine 



Figure 6 Adding the Peak Height Tool to Measure Limiting Current

Figure 7: Addition of Peak Height Tool to Measure Limiting Current
By dragging the control points on the tool around you can draw a proper baseline (see Figure 8).

Figure 8: A Proper Baseline for Measurement of the Limiting Current
The baseline type that is initially chosen typically does a good job for a one component reversible system such as that shown above. However, you can change the baseline type by right clicking on the tool and selecting “Properties” (see Figure 9).

Figure 9: Selection of Baseline Properties
This will bring up a dialog box where you can select the type of baseline from the drop-down menu (see Figure 10).

Figure 10: Dialog Box Showing Baseline Types
After all the limiting currents have been determined (see Figure 11) you can plot 


Figure 11: Results with Peak Heights for a Ferrocene Solution

Figure 12:
As determined by the equation shown below, plotting 



where 

Figure 13: Koutecky-Levich plot.
Theory
The theory section is split between what has already been defined for RDE in a prior webpage, and additional information about Koutecky-Levich RDE.
Rotating Disk
The following theoretical introduction to RDE is intended to give the reader a general understanding so that they may better understand what parameters affect the outcome in a typical experiment. A more detailed description can be found in Bard and Faulkner.2 Rotating the electrode is a method of forced convection with the purpose of continually delivering material to the electrode in a controlled manner. The rotating rod creates a vortex flow underneath the electrode which pulls material upwards.
The purpose of rotating the electrode is to keep the solution homogeneous. However, next to the electrode is a stagnant layer, called the Levich layer which actually “clings” to the electrode and rotates with it. Inside this layer, the primary mode of mass transport is diffusion. Even though the primary mode of mass transport is diffusion like in cyclic voltammetry, linear sweep voltammetry, or chronoamperometry the concentration gradient at the electrode remains constant with respect to time. Since the concentration gradient remains constant with respect to time, the current is a steady-state current.
The thickness of the Levich layer will depend upon the experimental conditions and is governed by the equation

where 
Table 1: Kinematic Viscosities for  Solutions at Solutions at  1 1 |
|
| Solution |  |
 |
 |
 |
 |
 (acetonitrile) (acetonitrile) |
 |
 (dimethylsulfoxide) (dimethylsulfoxide) |
 |
| Pyridine |  |
 (dimethylformamide) (dimethylformamide) |
 |
| N,N-Dimethylacetamide |  |
 (hexamethylphosphoramide) (hexamethylphosphoramide) |
 |
 |
 |
The limiting current at electrode is proportional to the thickness of the Levich layer and is defined by

where

Koutecky-Levich RDE
The limiting current described in the section above is the diffusion limited current for RDE. KL-RDE is used to extract heterogeneous rate constants for the species of interest. This is accomplished by extrapolating what the limiting current would be at infinitely high rotation speeds. Consider the equation below
where 




where 

Application
In the first example, Rajasekharan et al.3 use KL-RDE to extract heterogeneous rate constants for for the reduction of free chlorine and monochloramine. Studying these two chlorines is significant because they are the most commonly used drinking water disinfectants. The researchers found that the reduction of free chlorine proceeds twice as fast as monochloramine.
The second example Finklestein et al.4 use KL-RDE to investigate the oxidation of 
The third example uses KL-RDE in a slightly different way. Rather than measuring heterogeneous rate constants, Shigehara et al.5 used KL-RDE to investigate self-exchange rates for 

In the final example, Kundu et al.6 used KL-RDE to investigate the oxygen reduction reaction (ORR) using doped and undoped carbon nanotubes. Rather than extracting a rate constant the researchers used the slope of the Koutecky-Levich plot to obtain the number of electrons transferred during the ORR. Using doped carbon nanotubes, the researchers found that the number of electrons transferred during the reaction was 4, meaning that oxygen is reduced to 

References
1. He, P. Anal. Chem., 1995, 67, 986-992.
2. Faulkner, L. R.; Bard, A. J. Potential Sweep Methods, Electrochemical Methods: Fundamentals and Applications, 2nd ed.; Wiley: New Jersey, 2000; 226-260.
4. Finklestein, D. A.; Da Mota, N.; Cohen, J. L.; Abruna, H. D. J. Phys. Chem. C., ASAP
5. Shigehara, K.; Oyama, N.; Anson, F. C. Inorg. Chem., 1981, 20, 518-522.
Additional Resources
Linear Sweep Voltammetry (LSV)
This article is part of the AfterMath Data Organizer Electrochemistry Guide
Detailed Description
Linear sweep voltammetry is simply cyclic voltammetry without a Vertex Potential and reverse scan. Users are referred to the description on Cyclic voltammetry for more detail regarding instrument setup and acquisition. The basic setup will be discussed here. The setup panel for linear sweep voltammetry is as shown in Figure 1. The user can choose an Initial potential, Final potential and Sweep rate. Electrode range can be Auto or chosen by the user provided the range of currents for the experiments are known.

Figure 1: Basic setup for linear sweep voltammetry.
The Advanced, Ranges, and Post Experiment Conditions tabs are identical in setup to the Cyclic voltammetry tabs. Finally, a plot of the potential applied to the electrode is shown in Figure 2. Note that the flat portions at the beginning of the scan and the end of the scan are the Induction period and Relaxation periods, respectively. Details regarding these two periods are provided in separate wikis. After completion of the experiment and relaxation period, the post experiment idle conditions are applied to the cell.

Figure 2: Applied potential for linear sweep voltammetry.
Theory and Application
The user is referred to the section on Cyclic voltammetry for a discussion on theory.
Most applications of linear sweep voltammetry relate to cyclic voltammetry. However, here are a few examples where linear sweep voltammetry was applied.
In the first example,1 Cheng and coworkers used linear sweep voltammetry to examine direct methane production using a biocathode containing methanogens in either an electrochemical system or a microbial electrolysis cell by a process called electromethanogenesis. Since the production of methane from 
In another example,2 Wang and coworkers used linear sweep voltammetry to examine the release of inorganic ions and DNA from an ionorganic ion/DNA bilayer film. Performing linear sweep voltammetry simultaneously with surface plasmon resonance, the researchers were able to show that the film disassembled upon sweeping the potential to more negative values. This allowed the researchers to demonstrate the controlled release of DNA for gene-targeting therapy.
References
Additional Resources
Links: Electrochemist's Guide, AfterMath User's Guide, AfterMath Main Support Page
Related Techniques: Cyclic Voltammetry (CV), Staircase Voltammetry (SCV)
Experimental Considerations
This article is part of the AfterMath Data Organizer Electrochemistry Guide
Interdependency of the Potentiostat and Electrochemical Cell
One of the key aspects of electrochemical measurements is that the potentiostat and the electrochemical cell are part of the same electrical circuit. If something is wrong with the cell, then the cell can make the potentiostat appear to have a problem. Or, if something is wrong with the potentiostat, it can cause an unwanted change to occur within the cell. Other kinds of laboratory instrumentation, from the lowly digital balance to more complex spectrophotometers and chromatographic detectors, are relatively immune to changes in the nature of the sample being analyzed and are less likely to cause unwanted changes in the composition of the sample. But the successful user of a potentiostat must always be aware of the significant interdependence between the potentiostat and the sample and plan experiments accordingly.
Part of this awareness is carefully managing the sequence of events before, during, and after an electroanalytical measurement. The initial conditions imposed upon an electrochemical cell (such as the working electrode potential) as well as the instrument settings (such as the current range) can have a direct bearing on the observed results of an electrochemical measurement. Similarly, the continuity of cell control (or lack thereof) after a measurement concludes can directly influence the composition of the portions of the electrochemical cell immediately adjacent to the electrode(s). Indeed, the surface properties of the electrodes themselves can be unwittingly altered by improper cell control.
Minimizing Cell Change for a Successful Electroanalytical Measurement

A strategy for a successful electroanalytical measurement often begins with devising a way to connect the electrodes to the potentiostat without any appreciable changes occurring within the cell. In the jargon of the electroanalytical chemist, this might be as simple as initially poising the working electrode at a potential where no Faradaic current flows. Or, in the jargon of the corrosion scientist, it might be wise to start an experiment with the corrosion sample at or near OCP (Open Circuit Potential, the potential at which there is no current at the working electrode).
The AfterMath software gives you control of the idle conditions which prevail while you are making electrode connections and before an experiment begins. These “pre-experiment” idle conditions can be adjusted at any time while the potentiostat is not doing an experiment. You can choose to hold the working electrode at a given potential under potentiostatic control or alternately, at a given current (usually zero amperes) under galvanostatic control. You may also choose to hold the electrode at OCP if you wish.
Click here for more information about adjusting pre-experiment idle conditions.
The Induction Period
As part of the experimental specifications for an electrochemical technique, AfterMath allows you to specify a period of time during which the cell is allowed to come into equilibrium with the initial signal levels applied to the working electrode(s). The period of time, called the induction period, is brief by default (usually less than 3 seconds); however, you may increase the duration of the induction period or you may skip the induction period if you wish.
Click here for more information about setting up the induction period.