Hydrodynamic Voltammetry Theory
10.1 – Forced Convection
The current signal recorded during an electrochemical experiment is easily influenced or disturbed by the convection of various molecules and ions due to bulk movement of the solution. Proper interpretation of the current signal must accurately account for any contributions (desired or undesired) from solution convection. Thus, the control of solution movement is a critical part of any electrochemical experiment design, and the issue of convection cannot be ignored. Two opposing approaches are typically used to address the convection issue. At one extreme, an experiment can be conducted in a quiescent solution, so that convection makes little or no contribution to the observed current. The opposite extreme involves forced convection, where the solution is actively stirred or pumped in a controlled manner.
At first glance, it may seem that the simplest and most obvious way to account for convection is to try to eliminate it entirely by using a quiescent (non-moving) solution. This is the approach used in many popular electroanalytical techniques(1) (including cyclic voltammetry, chronoamperometry, square wave voltammetry, and differential pulse voltammetry). The timescale for these methods is generally less than 30 seconds, and on such short timescales, the influence of convection in an unstirred solution is generally negligible. On longer timescales, however, even unstirred solutions are prone to convective interference from thermal gradients and subtle environmental vibrations.
For long duration (steady-state) experiments, convection is unavoidable, so actively forcing(2) the solution to move in a well-defined and controlled manner is the preferred approach. An entire family of electroanalytical methods (broadly categorized as hydrodynamic voltammetry) couples precise control of solution flow with rigorous mathematical models defining the flow. Some of the many examples of hydrodynamic voltammetry include placing an electrode in a flow cell,(3) firing a jet of solution at an electrode target,(4-5) embedding an electrode in a microfluidic channel,(6) vibrating a wire-shaped electrode,(7) subjecting the solution to ultrasonication,(8) and rotating the electrode.(2,9-14)
By far the most popular and widely used hydrodynamic methods are those that involve a rotating electrode. The rotating electrode geometries most amenable to mathematical modeling are the rotating disk electrode (RDE),(9-14) the rotating ring-disk electrode (RRDE),(15-26) and the rotating cylinder electrode (RCE).(27-32) Researchers take advantage of the stable, steady-state laminar flow conditions adjacent to an RDE or RRDE to carefully gather information about electrode reaction kinetics.(13,14,21,26,33-43) In contrast, the relatively chaotic and turbulent conditions adjacent to an RCE are exploited by corrosion scientists(44-69) wishing to mimic flow-induced pipeline corrosion conditions in the laboratory. Development of the RDE and RRDE as routine analytical tools has largely been carried out by the community of academic electroanalytical chemists, while the RCE has primarily been a tool used by the corrosion and electroplating industries.
10.2 – Half Reactions
Regardless of the rotating electrode geometry being used, the common theme is that an ion or molecule is being conveyed to the electrode surface, and upon arrival, it is either oxidized or reduced depending upon the potential applied to the rotating electrode. If a sufficiently positive potential is applied to the electrode, then the molecules (or ions) tend to be oxidized, and conversely, if a sufficiently negative potential is applied to the rotating electrode, the molecules (or ions) tend to be reduced.
Reduction at a rotating electrode implies that electrons are being added to the ion or molecule by flowing out of the electrode and into the solution. A current travelling in this direction is said to be a cathodic current. The general form of a reduction half-reaction occurring at an electrode may be written as follows:

where 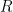 represents the reduced form of the molecule (or ion),
represents the reduced form of the molecule (or ion),  represents the oxidized form of the molecule (or ion), and
represents the oxidized form of the molecule (or ion), and  is the total number of electrons added to the molecule (or ion) when it is converted from the oxidized form () to the reduced form ().
is the total number of electrons added to the molecule (or ion) when it is converted from the oxidized form () to the reduced form ().
Oxidation at a rotating electrode implies that electrons are being removed from an ion or molecule and are travelling out of the solution and into the electrode. A current travelling in this direction is said to be an anodic current, and the oxidation occurring at the electrode can be represented by the following redox half reaction,

Given that electrochemical half reactions can occur in either direction, they are often written using chemical equilibrium notation* as follows:

Each half reaction has an associated standard electrode potential ( ) which is a thermodynamic quantity related to the free energy associated with the equilibrium. Like many other standard thermodynamic quantities, the standard electrode potential corresponds to a given standard state. The standard state corresponds to a thermodynamic system where the chemical activities of and are unity (i.e., when all solution concentrations are
) which is a thermodynamic quantity related to the free energy associated with the equilibrium. Like many other standard thermodynamic quantities, the standard electrode potential corresponds to a given standard state. The standard state corresponds to a thermodynamic system where the chemical activities of and are unity (i.e., when all solution concentrations are  , all gases are present at
, all gases are present at  partial pressure, and other materials are present as pure phases with unity activity).
partial pressure, and other materials are present as pure phases with unity activity).
To account for the (likely) possibility of non-unity activities, the Nernst equation (see below) can be used to express the equilibrium electrode potential ( ) in terms of the actual activities.
) in terms of the actual activities.
![E_{NERNSTIAN} = E^{\circ} + \frac{RT}{nF}ln\left[{\frac{{\alpha}_O}{{\alpha}_R}}\right]](https://s0.wp.com/latex.php?latex=E_%7BNERNSTIAN%7D+%3D+E%5E%7B%5Ccirc%7D+%2B+%5Cfrac%7BRT%7D%7BnF%7Dln%5Cleft%5B%7B%5Cfrac%7B%7B%5Calpha%7D_O%7D%7B%7B%5Calpha%7D_R%7D%7D%5Cright%5D&bg=ffffff&fg=000&s=3&c=20201002)
where  is the Faraday constant (
is the Faraday constant ( ), is the ideal gas constant (
), is the ideal gas constant ( ), and
), and  is the absolute temperature (
is the absolute temperature ( ). Usually, the activities of molecules or ions dissolved in solution are assumed to be the same as their molar concentrations, so the Nernst Equation is often written as follows
). Usually, the activities of molecules or ions dissolved in solution are assumed to be the same as their molar concentrations, so the Nernst Equation is often written as follows
![E_{NERNSTIAN} = E^{\circ} + \frac{RT}{nF}ln\left[{\frac{C_O}{C_R}}\right]](https://s0.wp.com/latex.php?latex=E_%7BNERNSTIAN%7D+%3D+E%5E%7B%5Ccirc%7D+%2B+%5Cfrac%7BRT%7D%7BnF%7Dln%5Cleft%5B%7B%5Cfrac%7BC_O%7D%7BC_R%7D%7D%5Cright%5D&bg=ffffff&fg=000&s=3&c=20201002)
where  and
and  are the concentrations of the dissolved molecules or ions in the oxidized and reduced forms, respectively, at the surface of the electrode. Note that any liquid or solid phase materials at the electrode surface (such as the solvent or the electrode itself) have unity activity and thus do not appear in the Nernst equation.
are the concentrations of the dissolved molecules or ions in the oxidized and reduced forms, respectively, at the surface of the electrode. Note that any liquid or solid phase materials at the electrode surface (such as the solvent or the electrode itself) have unity activity and thus do not appear in the Nernst equation.
This half reaction at an electrode can be driven in the cathodic (reducing) direction by applying a potential to the electrode ( ) which is more negative than the equilibrium electrode potential (
) which is more negative than the equilibrium electrode potential ( ). Conversely, the half reaction can be driven in the oxidizing (anodic) direction by applying a potential more positive than the equilibrium electrode potential (
). Conversely, the half reaction can be driven in the oxidizing (anodic) direction by applying a potential more positive than the equilibrium electrode potential ( ).
).
—–
* By convention, redox half reactions are generally tabulated in textbooks and other reference works as reduction reactions (with the oxidized form on the left side and the reduced form on the right side, as shown above), but it is understood that the reaction may occur in either direction depending upon the potential applied to the electrode.
10.3 – Voltammetry
The term voltammetry refers broadly to any method where the electrode potential is varied while the current is measured.(1-2) The terminology associated with voltammetry varies across different industries and academic disciplines, but the underlying principles of all voltammetric techniques are very similar.
The most common form of voltammetry involves sweeping the electrode potential from an initial value to a final value at a constant rate. When working in the context of electroanalytical chemistry with a non-rotating electrode, this technique is called linear sweep voltammetry (LSV). In the context of corrosion science, this kind of technique is usually called linear polarization resistance (LPR) or a Tafel analysis. The term cyclic voltammetry (CV) refers to a method where the electrode potential is swept repeatedly back-and-forth between two extremes.
When working with a rotating electrode, it is common to further specify the kind of electrode being used as part of the technique name, such as rotating disk voltammetry, rotating ring-disk voltammetry, or rotating cylinder voltammetry. In each of these techniques, the rotation rate is held constant as the electrode is swept from one potential to another potential at a constant sweep rate. In electroanalytical chemistry, the potential sweep usually spans at least  on either side of the standard electrode potential, and rotation rates are usually between
on either side of the standard electrode potential, and rotation rates are usually between  and
and  . However, in the context of a corrosion study, the potential sweep may span a much narrower range (
. However, in the context of a corrosion study, the potential sweep may span a much narrower range ( ) using a slower sweep rate (less than
) using a slower sweep rate (less than  ) with an emphasis on higher rotation rates.
) with an emphasis on higher rotation rates.
As an example, consider a solution that initially contains only the oxidized form of a molecule or ion. A rotating electrode is placed in this solution and is initially poised at a potential that is more positive than the standard potential. At this potential, there is little or no current because there is nothing to oxidize (the molecule or ion is already oxidized), and the potential is not (yet) negative enough to cause any appreciable reduction of the molecule or ion.
Next, the electrode potential is slowly ( ) swept in the negative (cathodic) direction (see Figure 10.1, left). As the applied potential approaches the standard electrode potential, a cathodic current is observed (see Figure 10.1, right). The cathodic current continues to increase as the potential moves past the standard electrode potential towards more negative potentials.
) swept in the negative (cathodic) direction (see Figure 10.1, left). As the applied potential approaches the standard electrode potential, a cathodic current is observed (see Figure 10.1, right). The cathodic current continues to increase as the potential moves past the standard electrode potential towards more negative potentials.
The current eventually reaches a maximum value (limiting current) once the applied potential is sufficiently negative relative to the standard electrode potential. At such a negative potential, any oxidized form of the molecule or ion () that reaches the surface of the electrode is immediately converted to the reduced form () as shown below.
The observed cathodic current is the result of electrons flowing out of the electrode and into the solution. The rate of electron flow is limited only by how fast the oxidized form () can arrive at the electrode surface. The maximum current observed in this circumstance is called the cathodic limiting current ( ).
).
Whenever an observed current is limited only by the rate at which material arrives at the electrode surface, the current is said to be mass transport limited. When working with a rotating electrode, the rate of mass transport is related to the rotation rate of the electrode. Rotating the electrode at a faster rate increases the rate at which material arrives at the electrode surface. Thus, the limiting current increases with increasing rotation rate. Experiments involving a rotating electrode are designed to purposefully exploit this fundamental relationship between the rotation rate and the limiting current.
Figure 10.1: Response to a Potential Sweep (Cathodic) from a Solution Initially Containing only the Oxidized Form ( ) with no Reduced Form (
) with no Reduced Form ( )
)
The cathodic sweep experiment described above (see Figure 10.1) applies to the case where the solution initially contains only the oxidized form () of the molecule or ion being studied. The opposite case yields similar results. Consider a solution that initially contains only the reduced form () of the molecule or ion being studied. The rotating electrode is initially poised at a potential that is about more negative than the standard potential. At this potential, there is little or no current because there is nothing to reduce (the molecule or ion is already reduced), and the potential is not (yet) positive enough to cause any appreciable oxidation of the molecule or ion.
Next, the electrode potential is slowly swept in the positive (anodic) direction (see Figure 10.2, left) and an anodic current is observed (see Figure 10.2, right). The anodic current eventually reaches a maximum value when the potential is sufficiently positive relative to the standard electrode potential. At this point, any of the reduced form () that reaches the electrode surface is immediately converted to the oxidized form ().

The observed current is the result of electrons flowing into the electrode. The maximum current observed is called the anodic limiting current ( ).
).
Figure 10.2: Response to a Potential Sweep (Anodic) from a Solution Initially Containing only the Reduced Form () with no Oxidized Form ()
10.3.1 – Voltammogram Plotting Conventions
The two streams of data recorded during a voltammetry experiment are the potential vs. time and the current vs. time. Rather than plot these two streams separately (as shown in Figure 10.3, top), it is more common to plot current vs. potential (as shown in Figure 10.3, bottom). Such a plot is called a voltammogram.
Figure 10.3: A Voltammogram is a Plot of Current versus Potential
Although most electroanalytical researchers agree that current should be plotted along the vertical axis and potential should be plotted along the horizontal axis, there is not widespread agreement as to the orientation (direction) for each axis. Some researchers plot positive (anodic, oxidizing) potentials toward the right while others plot negative (cathodic, reducing) potential toward the right (as per classical polarography tradition). Furthermore, some researchers plot anodic (oxidizing) current upward along the vertical axis, while others plot cathodic (reducing) current in the upward direction.
This means there are four possible conventions for plotting a voltammogram, and one should always take a moment to ascertain the orientation of the axes before interpreting a voltammogram. Fortunately, of the four possible ways to plot a voltammogram, only two are commonly used. The older tradition (based on classical polarography) plots cathodic current upwards along the vertical axis and negative (cathodic, reducing) potentials toward the right along the horizontal axis. A complex voltammogram involving four different limiting currents (see Figure 10.4, left) illustrates this convention, which is sometimes called the “North American” convention.
North American Convention

European Convention

Figure 10.4: Two Popular Voltammogram Plotting Conventions
The same data may be plotted using the “European” convention (see Figure 10.4, right). This convention plots anodic currents upward along the vertical axis and more positive (anodic, oxidizing) potentials to the right along the horizontal axis. The European convention is more readily understood by those outside the electroanalytical research community (because positive values are plotted to the right along the horizontal axis).
The European convention is used throughout the remainder of this article. Note that this choice also implies a mathematical sign convention for the current. Specifically, positive current values are considered anodic, and negative current values are considered cathodic in this document. This sign convention is somewhat arbitrary, and electrochemical data processing software available from various manufacturers may or may not use this sign convention.
10.3.2 – Measuring Limiting Currents
The theoretical voltammetric response from a rotating electrode is a symmetric sigmoid-shaped wave (like the ideal voltammograms shown in Figure 10.3 and Figure 10.4). A perfect sigmoid has a flat baseline current before the wave and a flat limiting current plateau after the wave. The height of the wave (as measured from the baseline current to the limiting current plateau) is the mass-transport limited current.
In actual “real world” experiments, the wave may be observed on top of a background current, and furthermore, the background current may be slightly sloped (see Figure 10.5). This (undesired) background current may be due to interference from oxidation or reduction of impurities or of the solvent itself. The background current may also be due to capacitive charging and discharging of the ionic double-layer that forms next to the polarized electrode surface.
Figure 10.5: Sloping Backgrounds in Voltammograms
When attempting to measure the (desired) Faradaic mass-transport limited current at a rotating electrode, it is often necessary to account for the (undesired, possibly sloping) background current. If the background current has a constant slope across the entire voltammogram, then it is fairly easy to extrapolate the sloping baseline to a point underneath the limiting current plateau (see Figure 10.5, left). The limiting current is measured as the (vertical) distance between the plateau and the extrapolated baseline. In voltammograms where there is more than one wave, the plateau for the first wave is used as the baseline for the second wave (see  in Figure 10.5, left).
in Figure 10.5, left).
In some cases, the slope of the background current is not constant across the entire voltammogram. That is, the slope of the baseline leading up to the wave can be different than the slope of the plateau after the wave. It can be very difficult to discern exactly where to measure the limiting current along such a voltammogram. One approach is to extrapolate the baseline forward through the wave and also extrapolate the plateau backward through the wave. Then, the limiting current is measured as the vertical distance between the baseline and plateau at a point corresponding to the center of the voltammogram (see in Figure 10.5, right).
Finally, it should be noted that when the oxidized form () and the reduced form () of a molecule or ion are both present in a solution at the same time, the voltammogram is likely to exhibit both a cathodic and an anodic limiting current (see Figure 10.6). It can be very difficult to measure the limiting current properly in this case, especially if there is also a sloping background current. For this reason, most experiments with rotating electrodes are conducted in solutions where only one form of the molecule or ion is initially present.
Figure 10.6: Voltammogram for a Solution Containing Both  and
and 
10.4 – Rotating Disk Electrode (RDE) Theory
The general theory describing mass transport at a rotating disk electrode (RDE) was developed by Benjamin Levich at the Institute of Electrochemistry at the Academy of Sciences of the USSR. Levich described the theory in his landmark book, Physicochemical Hydrodynamics, originally published in Russian in 1952. Ten years later, Levich’s book was translated(9) from Russian to English, and the RDE became more widely known(11) to western researchers. In the early 1960’s, Stanley Bruckenstein(10) at the University of Minnesota (and his students Dennis Johnson and Duane Napp) and Ronnie Bell(12) at Oxford University (and his student John Albery) began working with rotating electrodes. Subsequent generations of researchers expanded on this initial work, and the rotating disk electrode has since grown into a mature tool for probing electrochemical reaction kinetics.(13)
The laminar flow at a rotating disk electrode conveys a steady stream of material from the bulk solution to the electrode surface. While the bulk solution far away from the electrode remains well-stirred by the convection induced by rotation, the portion of the solution nearer to the electrode surface tends to rotate with the electrode. Thus, if the solution is viewed from the frame of reference of the rotating electrode surface, then the solution appears relatively stagnant. This relatively stagnant layer is known as the hydrodynamic boundary layer, and its thickness ( ) can be approximated,
) can be approximated,

in terms of the kinematic viscosity of the solution ( ) and the angular rotation rate (
) and the angular rotation rate ( ), where f is the rotation rate in revolutions per minute). In an aqueous solution at a moderate rotation rate (
), where f is the rotation rate in revolutions per minute). In an aqueous solution at a moderate rotation rate ( ), the stagnant layer is approximately
), the stagnant layer is approximately  to
to  thick.
thick.
Net movement of material to the electrode surface can be described mathematically by applying general convection-diffusion concepts from fluid dynamics. Mass transport of material from the bulk solution into the stagnant layer occurs by convection (due to the stirring action of the rotating electrode). But after the material enters the stagnant layer and moves closer to the electrode surface, convection becomes less important and diffusion becomes more important. Indeed, the final movement of an ion or molecule to the electrode surface is dominated by diffusion across a very thin layer of solution immediately adjacent to the electrode known as the diffusion layer.
The diffusion layer is much thinner than the hydrodynamic layer. The diffusion layer thickness ( ) can be approximated as follows,
) can be approximated as follows,

in terms of the diffusion coefficient ( ) of the molecule or ion. For a molecule or ion with a typical diffusion coefficient (
) of the molecule or ion. For a molecule or ion with a typical diffusion coefficient ( ) in an aqueous solution, the diffusion layer is about twenty times thinner than the stagnant layer (
) in an aqueous solution, the diffusion layer is about twenty times thinner than the stagnant layer ( ).
).
The first mathematical treatment of convection and diffusion towards a rotating disk electrode was given by Levich. Considering the case where only the oxidized form of a molecule (or ion) of interest is initially present in the electrochemical cell, the cathodic limiting current () observed at a rotating disk electrode is given by the Levich equation,(2,9)

in terms of the concentration () of the oxidized form in the solution, the Faraday constant ( ), the electrode area (
), the electrode area (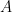 ), the kinematic viscosity of the solution (), the diffusion coefficient (
), the kinematic viscosity of the solution (), the diffusion coefficient (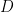 ) of the oxidized form, and the angular rotation rate (
) of the oxidized form, and the angular rotation rate ( ). Alternatively, when the solution initially contains only the reduced form, the Levich equation for the anodic limiting current () can be written as
). Alternatively, when the solution initially contains only the reduced form, the Levich equation for the anodic limiting current () can be written as

where the concentration term () is for the reduced form rather than the oxidized form.
10.4.1 – Levich Study
A Levich Study is a common experiment performed using a rotating disk electrode in which a series of voltammograms is acquired over a range of different rotation rates. For a simple electrochemical system where the rate of the half reaction is governed only by mass transport to the electrode surface, the overall magnitude of the voltammogram should increase with the square root of the rotation rate (see Figure 10.7, left).
Figure 10.7: Levich Study – Voltammograms at Various Rotation Rates
The currents measured during a Levich study are usually plotted against the square root of the rotation rate on a graph called a Levich plot. As predicted by the Levich equation, the limiting current (see red circles on Figure 10.7, right) increases linearly with the square root of the rotation rate (with a slope of  ) and the line intercepts the vertical axis at zero. It is common to choose a set of rotation rates that are multiples of perfect squares (such as
) and the line intercepts the vertical axis at zero. It is common to choose a set of rotation rates that are multiples of perfect squares (such as  ,
,  ,
,  ,
,  , etc.) to facilitate construction of this plot.
, etc.) to facilitate construction of this plot.
If the electrochemical half-reaction observed during a Levich study is a simple and reversible half reaction (with no complications due to sluggish kinetics or coupled chemical reactions), then the shapes of the mass-transport controlled voltammograms will be sigmoidal regardless of the rotation rate. This means that the current observed at any given potential along the voltammogram will vary linearly with the square root of the rotation rate (see Figure 10.7, right). But, it is important to remember that the Levich equation only applies to the limiting current, not to the currents along the rising portion of the sigmoid.
Limiting Current

Levich Plot

Koutecky-Levich Plot

Figure 10.8: Levich Study – Limiting Current versus Rotation Rate
Because the Levich equation only applies to the limiting current, the results from a Levich experiment are typically presented as a simple plot of the limiting current versus the square root of the rotation rate (see Figure 10.8, center). An alternate method of presenting the data from a Levich study is based on a rearrangement of the Levich equation in terms of the reciprocal current.

A plot of reciprocal current versus the reciprocal square root of the angular rotation rate (see Figure 10.8, right) is called a Koutecky-Levich(2,14) plot. Again, for a simple and reversible half reaction with no complications the data fall along a straight line that intercepts the vertical axis at zero. If the line intercepts the vertical axis above zero, however, this is a strong indication that the half-reaction is limited by sluggish kinetics rather than by mass transport.
10.4.2 – Koutecky-Levich Analysis
When the rate of a half reaction occurring at an electrode surface is limited by a combination of mass transport and sluggish kinetics, it is often possible to use a rotating disk electrode to elucidate both the mass transport parameters (such as the diffusion coefficient) and the kinetic parameters (such as the standard rate constant,  ) from a properly designed Levich study. A full treatment of this kind of analysis(14) is beyond the scope of this document, but the following is a general description of how to extract kinetic information from a set of rotating disk voltammograms.
) from a properly designed Levich study. A full treatment of this kind of analysis(14) is beyond the scope of this document, but the following is a general description of how to extract kinetic information from a set of rotating disk voltammograms.
When the electron transfer process at an electrode surface exhibits sluggish kinetics, the voltammogram appears stretched out along the potential axis and the shape of the sigmoidal wave is slightly distorted. Comparing a set of voltammograms with facile kinetics (see Figure 10.7) with a set of voltammograms with sluggish kinetics (see Figure 10.9), the mass transport limited current plateau (marked by red circles in each figure) is shifted further away from the standard electrode potential () when there are slow kinetics. Stated another way, when a sluggish redox half reaction is studied with a rotating disk electrode, a larger overpotential must be applied to the electrode to overcome the sluggish kinetics and reach the mass transport limited current.
Figure 10.9: Koutecky Levich Study – Voltammograms Revealing Sluggish Kinetics
This distortion of the ideal sigmoidal shape of the voltammogram can be exploited as a way to measure the standard rate constant (). The general approach is to acquire a set of voltammograms at different rotation rates (i.e., perform a Levich study) and then plot the reciprocal current (sampled at particular locations along the rising portion of each voltammogram) on a Koutecky-Levich Plot. In the example provided (see Figure 10.9, left), the current was sampled at two locations along the rising portion of the voltammograms (at  and
and  , marked with blue triangles and purple squares) and at one location on the limiting current plateau (at
, marked with blue triangles and purple squares) and at one location on the limiting current plateau (at  , marked with red circles). A linear relationship is evident (see Figure 10.9, right) when these sampled currents are plotted on a Koutecky-Levich plot.
, marked with red circles). A linear relationship is evident (see Figure 10.9, right) when these sampled currents are plotted on a Koutecky-Levich plot.
For the set of currents sampled on the limiting current plateau (red circles), an extrapolation back to the vertical axis (i.e., to infinite rotation rate) yields a zero intercept. This is the identical result obtained for a facile half-reaction (see Figure 10.8, right) because these currents are sampled at a high enough overpotential that there are no kinetic limitations. Only mass transport limits the current, and the usual Levich behavior applies.
However, for the two sets of currents sampled on the rising portion of the voltammogram (see Figure 10.9, blue triangles and purple squares), the extrapolation back to the vertical axis yields non-zero intercepts. This non-zero intercept indicates a kinetic limitation, meaning that even if mass transport were infinite (i.e., infinite rotation rate), the rate of the half-reaction would still be limited by the slow kinetics at the electrode surface.
The linear portion of the data on a Koutecky-Levich plot is described by the Koutecky-Levich equation (below).

Plotting the reciprocal current ( ) against the reciprocal square root of the angular rotation rate (
) against the reciprocal square root of the angular rotation rate ( ) yields a straight line with an intercept equal to the reciprocal kinetic current (
) yields a straight line with an intercept equal to the reciprocal kinetic current ( ). The kinetic current is the current that would be observed in the absence of any mass transport limitations. By measuring the kinetic current at a variety of different overpotentials along the voltammogram, it is possible to determine the standard rate constant for the electrochemical half reaction.
). The kinetic current is the current that would be observed in the absence of any mass transport limitations. By measuring the kinetic current at a variety of different overpotentials along the voltammogram, it is possible to determine the standard rate constant for the electrochemical half reaction.
Further details regarding Koutecky-Levich theory, including various forms of the Koutecky-Levich equation which pertain to different electrochemical processes, can be found in the literature.(14)
10.5 – Rotating Ring-Disk Electrode (RRDE) Theory
In 1958, Russian electrochemist Alexander Frumkin suggested the idea of placing a concentric ring electrode around the rotating disk electrode.(15) His colleague, Lev Nekrasov, supervised construction of the world’s first rotating ring-disk electrode (RRDE) apparatus.(16-19) At the same time, Benjamin Levich and Yuri Ivanov began working on a theoretical description of solution flow at the RRDE. The four Russian researchers published their initial findings in 1959, and their work caught the attention of both Stanley Bruckenstein at the University of Minnesota and John Albery at Oxford University. Bruckenstein travelled to Moscow to learn more about the RRDE,(20) and after he returned home in 1965, Albery joined Bruckenstein’s research group (along with Dennis Johnson and Duane Napp). The experimental and mathematical work performed by these four researchers at Minnesota generated a significant series of papers about the RRDE(21-26) and placed the new technique on a firm theoretical foundation. Albery returned to Oxford and (working with his student Michael Hitchman) drew these theoretical papers together in a seminal volume titled Ring-Disc Electrodes.(21)
The overall flow pattern at the RRDE initially brings molecules and ions to the disk electrode. After encountering the disk electrode, the subsequent outward radial flow carries a fraction of these molecules or ions past the surface of the ring electrode. This flow pattern allows products generated (upstream) by the half reaction at the disk electrode to be detected as they are swept (downstream) past the ring electrode.
Two of the key parameters which characterize a given ring-disk geometry are the collection efficiency(23) and the transit time. The collection efficiency is the fraction of the material from the disk which subsequently flows past the ring electrode. It can be expressed as a fraction between  and
and  or as a percentage. Typical ring-disk geometries have collection efficiencies between
or as a percentage. Typical ring-disk geometries have collection efficiencies between  and
and  . The transit time is a more general concept indicating the average time required for material at the disk electrode to travel across the gap between the disk and the ring electrode. Obviously, the transit time is a function of both the gap distance and the rotation rate.
. The transit time is a more general concept indicating the average time required for material at the disk electrode to travel across the gap between the disk and the ring electrode. Obviously, the transit time is a function of both the gap distance and the rotation rate.
10.5.1 – Theoretical Computation of the Collection Efficiency
The theoretical collection efficiency can be computed(2,23) from the three principle diameters describing the RRDE geometry: the disk outer diameter ( ), the ring inner diameter (
), the ring inner diameter ( ), and the ring outer diameter (
), and the ring outer diameter ( ). This somewhat tedious computation is made easier by normalizing the ring diameters with respect to the disk diameter as follows:
). This somewhat tedious computation is made easier by normalizing the ring diameters with respect to the disk diameter as follows:


Three additional quantities are defined in terms of the normalized diameters as follows:



If a complex function,  , is defined as follows,
, is defined as follows,
![G(x) = \frac{1}{4} + \left(\frac{\sqrt{3}}{4{\pi}}\right) ln \left[{\frac{(x^{1/3} + 1)^3}{x+1}}\right] + \frac{3}{2{\pi}} {\arctan}\left[{\frac{2x^{1/3}-1}{\sqrt{3}}}\right]](https://s0.wp.com/latex.php?latex=G%28x%29+%3D+%5Cfrac%7B1%7D%7B4%7D+%2B+%5Cleft%28%5Cfrac%7B%5Csqrt%7B3%7D%7D%7B4%7B%5Cpi%7D%7D%5Cright%29+ln+%5Cleft%5B%7B%5Cfrac%7B%28x%5E%7B1%2F3%7D+%2B+1%29%5E3%7D%7Bx%2B1%7D%7D%5Cright%5D+%2B+%5Cfrac%7B3%7D%7B2%7B%5Cpi%7D%7D+%7B%5Carctan%7D%5Cleft%5B%7B%5Cfrac%7B2x%5E%7B1%2F3%7D-1%7D%7B%5Csqrt%7B3%7D%7D%7D%5Cright%5D&bg=ffffff&fg=000&s=3&c=20201002)
then the theoretical collection efficiency ( ) for a rotating ring disk electrode is given by the following equation:
) for a rotating ring disk electrode is given by the following equation:

10.5.2 – Empirical Measurement of the Collection Efficiency
Direct computation of the theoretical collection efficiency is possible using the above relationships if the actual machined dimensions of the disk and ring are known for a particular RRDE. In practice, the actual RRDE dimensions may not be known due to uncertainties in the machining process and changes in the dimensions induced by electrode polishing or temperature cycling. For this reason, it is common practice to empirically measure the collection efficiency using a well-behaved redox system rather than to rely upon a computed value.
The ferrocyanide/ferricyanide half reaction is a simple, single-electron, reversible half reaction that is often used as the basis for measuring collection efficiency.(36) The RRDE is placed in a solution containing a small concentration ( ) of potassium ferricyanide,
) of potassium ferricyanide,  , in a suitable aqueous electrolyte solution (such as
, in a suitable aqueous electrolyte solution (such as  potassium nitrate,
potassium nitrate,  ) and is operated at rotation rates between
) and is operated at rotation rates between  and
and  . Initially, both the ring and the disk electrodes are held at a sufficiently positive potential that no reaction occurs. Then, the potential of the disk electrode is slowly swept (
. Initially, both the ring and the disk electrodes are held at a sufficiently positive potential that no reaction occurs. Then, the potential of the disk electrode is slowly swept ( ) towards more negative potentials, and a cathodic current is observed which corresponds to the reduction of ferricyanide to ferrocyanide at the disk.
) towards more negative potentials, and a cathodic current is observed which corresponds to the reduction of ferricyanide to ferrocyanide at the disk.
(reduction of ferricyanide to ferrocyanide at disk)
As ferricyanide is reduced at the disk electrode, the ferrocyanide generated by this process is swept outward (radially) away from the disk electrode and toward the ring electrode. The ring electrode is held constant at a positive (oxidizing) potential throughout the experiment. Some (but not all) of the ferrocyanide generated at the disk travels close enough to the ring electrode that it is oxidized back to ferricyanide. Thus, an anodic current is observed at the ring electrode due to the oxidation of ferrocyanide to ferricyanide at the ring.
(oxidation of ferrocyanide to ferricyanide at ring)
The measured ratio of the ring (anodic) limiting current to the disk (cathodic) limiting current is the empirical collection efficiency. As the rotation rate increases, both the disk and the ring currents increase (see Figure 10.10). Because both the anodic and cathodic limiting currents are proportional to the square root of the rotation rate, the empirical collection efficiency is expected to be independent of the rotation rate.
Figure 10.10: Rotating Ring-Disk Voltammograms at Various Rotation Rates
Once the collection efficiency value has been established empirically for a particular RRDE, it can be treated as a property of that particular RRDE, even if the RRDE is used to study a different half reaction in a different solution on a different day. Although the empirically measured collection efficiency ( ) is a ratio of two currents with opposite mathematical signs (anodic and cathodic), the collection efficiency is always expressed as a positive number.
) is a ratio of two currents with opposite mathematical signs (anodic and cathodic), the collection efficiency is always expressed as a positive number.

10.5.3 – Generator/Collector Experiments
When a molecule or ion is oxidized or reduced at an electrode, it is often transformed into an unstable intermediate chemical species which, in turn, is likely to undergo additional chemical changes. The intermediate may have a long enough lifetime that it is capable of moving to the ring electrode and being detected. Or, the intermediate may be so unstable that it decays away before it can be detected at the ring. Consider the following reaction scheme at a rotating ring-disk electrode:
(reduction of A to unstable intermediate X at disk electrode)
(chemical decay of X to electrochemically inactive Z)
(oxidation of X back to A at ring electrode)
In the above scheme, the disk electrode is poised at a potential where is reduced to  , and the cathodic limiting current observed at the disk (
, and the cathodic limiting current observed at the disk ( ) is a measure of how much is being “generated” at the disk electrode. At the same time, the ring electrode is poised at a more positive potential where is oxidized back to , and the anodic limiting current observed at the ring (
) is a measure of how much is being “generated” at the disk electrode. At the same time, the ring electrode is poised at a more positive potential where is oxidized back to , and the anodic limiting current observed at the ring ( ) is a measure of much is being “collected” at the ring. There is also a competing chemical reaction which is capable of eliminating before it has a chance to travel from the disk to the ring.
) is a measure of much is being “collected” at the ring. There is also a competing chemical reaction which is capable of eliminating before it has a chance to travel from the disk to the ring.
The ratio of the ring current to the disk current under these conditions is called the apparent collection efficiency ( ).
).

By comparing the apparent collection efficiency () to the previously measured empirical collection efficiency () for the same RRDE, it is possible to deduce the rate at which the competing chemical pathway is converting to  . That is, it is possible to use an RRDE “generator/collector” experiment to measure the kinetic behavior of unstable electrochemical intermediates.
. That is, it is possible to use an RRDE “generator/collector” experiment to measure the kinetic behavior of unstable electrochemical intermediates.
Whenever is equal to , it is an indication that the decay rate of the intermediate (via the  pathway) is small with respect to the transit time required for to travel from the disk to the ring. One way to shorten the transit time is to spin the RRDE at a faster rate. At high rotation rates, the apparent collection efficiency should approach the empirical collection efficiency. Conversely, at slower rotation rates, the apparent collection efficiency may be smaller ( < ) because some of the intermediate is consumed by the competing chemical pathway before can travel to the ring.
pathway) is small with respect to the transit time required for to travel from the disk to the ring. One way to shorten the transit time is to spin the RRDE at a faster rate. At high rotation rates, the apparent collection efficiency should approach the empirical collection efficiency. Conversely, at slower rotation rates, the apparent collection efficiency may be smaller ( < ) because some of the intermediate is consumed by the competing chemical pathway before can travel to the ring.
By recording a series of rotating ring-disk voltammograms at different rotation rates and analyzing the results, it is possible to estimate the rate constant (k) associated with the intermediate chemical decay pathway. Various relationships have been proposed for this kind of analysis,(2) and one of the simplest is shown below.

A plot of the ratio of the empirical to the apparent collection efficiency versus the reciprocal angular rotation rate should be linear. The slope of such a plot can yield the rate constant if the kinematic viscosity ( ) and the diffusion coefficient () are known.
) and the diffusion coefficient () are known.
10.5.4 – Comparing Two Competing Pathways
Sometimes the intermediate generated by an electrochemical process can decay via two different pathways. As long as one of these pathways leads to an electrochemically active chemical species that can be detected at the ring, it is possible to determine which decay pathway is favored. Consider the following scheme:
In the above scheme, the disk electrode is poised at a potential where is reduced to , and the cathodic limiting current observed at the disk () is a measure of how much is being “generated” at the disk electrode. The intermediate is unstable, and as it is swept away from the disk and toward the ring, it rapidly decays to either  or . By the time these species reach the ring, all of the has decayed away, and the solution in contact with the ring contains both and . The species is electrochemically inactive and cannot be detected by the ring, but the species is active. By carefully poising the ring electrode at a potential appropriate for detecting (in this case, by oxidizing to
or . By the time these species reach the ring, all of the has decayed away, and the solution in contact with the ring contains both and . The species is electrochemically inactive and cannot be detected by the ring, but the species is active. By carefully poising the ring electrode at a potential appropriate for detecting (in this case, by oxidizing to  ), it is possible for the ring to “collect” any which arrives at the surface of the ring.
), it is possible for the ring to “collect” any which arrives at the surface of the ring.
The ratio of the ring current (due to being detected at the ring) to the disk current (due to being generated at the disk) reveals the extent to which the  pathway is favored in comparison to the pathway. The fraction of the decay by the pathway (
pathway is favored in comparison to the pathway. The fraction of the decay by the pathway ( ) can be computed as follows.
) can be computed as follows.

Note in the above equation that the fraction ( ) carefully accounts for any difference in the number of electrons involved in the disk half reaction and the number of electrons involved when detecting at the ring electrode. Schemes involving more complex stoichiometry may require additional correction factors.
) carefully accounts for any difference in the number of electrons involved in the disk half reaction and the number of electrons involved when detecting at the ring electrode. Schemes involving more complex stoichiometry may require additional correction factors.
The most commonly studied reaction at the RRDE is undoubtedly the oxygen reduction reaction (ORR).(33-43) When oxygen ( ) is dissolved in acidic media and reduced at a platinum electrode, one pathway leads to water as the ultimate reduction product while the other pathway leads to the formation of peroxide anions. In the context of hydrogen fuel cell research, the pathway which leads to water is preferred, and it is commonly called the four-electron pathway. The path to peroxide formation is called the two-electron pathway, and it is undesirable for a number of reasons, including the fact that peroxide can damage various polymer membrane materials found in a fuel cell. Further details on how to use an RRDE “generator/collector” experiment to distinguish between the two-electron and four-electron ORR pathways can be found in the electrochemical literature.(33,36)
) is dissolved in acidic media and reduced at a platinum electrode, one pathway leads to water as the ultimate reduction product while the other pathway leads to the formation of peroxide anions. In the context of hydrogen fuel cell research, the pathway which leads to water is preferred, and it is commonly called the four-electron pathway. The path to peroxide formation is called the two-electron pathway, and it is undesirable for a number of reasons, including the fact that peroxide can damage various polymer membrane materials found in a fuel cell. Further details on how to use an RRDE “generator/collector” experiment to distinguish between the two-electron and four-electron ORR pathways can be found in the electrochemical literature.(33,36)
10.6 – Rotating Cylinder Electrode (RCE) Theory
The rotating disk and ring-disk electrodes were developed primarily as a result of academic electroanalytical chemistry research. In contrast, the theory for the rotating cylinder electrode (RCE) was developed by industrial researchers(44-46) in the corrosion and electroplating communities. While the flow of solution at a rotating disk (or ring-disk) is laminar over a wide range of rotation rates, the flow at the surface of a rotating cylinder is turbulent(31) at all but the slowest rotation rates. Thus, the RCE is an excellent tool for creating and controlling turbulent flow conditions in the laboratory, and it is most commonly used to mimic turbulent corrosion conditions found in large scale industrial settings such as oilfield pipeline corrosion.(56-69)
The turbulent flow at a rotating cylinder electrode conveys material from the bulk solution towards the electrode surface. While the bulk solution remains well stirred by the main vortex induced by the rotating electrode, the layer of solution adjacent to the cylinder surface tends to rotate with the electrode. Thus, a high shear condition is set up at the surface of the rotating cylinder, spinning off smaller Taylor vortices adjacent to the rotating electrode.
Net movement of material to the surface of a rotating cylinder was first characterized by Morris Eisenberg(27,28) in 1954 (about the same time that Levich was describing the rotating disk electrode). Eisenberg’s work eventually led to the Eisenberg equation which gives the limiting current at a rotating cylinder electrode

in terms of the concentration ( ) and diffusion coefficient () of the molecule or ion being studied, the Faraday constant (), the electrode area (), the diameter of the cylinder (
) and diffusion coefficient () of the molecule or ion being studied, the Faraday constant (), the electrode area (), the diameter of the cylinder ( ), the kinematic viscosity of the solution (
), the kinematic viscosity of the solution ( ), and the angular rotation rate (
), and the angular rotation rate ( , where
, where  is the rotation rate in revolutions per minute). In the years since Eisenberg’s initial work with the rotating cylinder, additional work by Gabe, Kear, Walsh, and Silverman has described industrial applications of the RCE.(29-32, 44-69)
is the rotation rate in revolutions per minute). In the years since Eisenberg’s initial work with the rotating cylinder, additional work by Gabe, Kear, Walsh, and Silverman has described industrial applications of the RCE.(29-32, 44-69)















































 Related Hardware Links:
Related Hardware Links: 



 (
( ) outer diameter. When properly mounted in the rotator, the upper
) outer diameter. When properly mounted in the rotator, the upper  (
( ) of the shaft is inside the motor unit, while the remaining length of the shaft extends down below the motor unit.
) of the shaft is inside the motor unit, while the remaining length of the shaft extends down below the motor unit.



 , provided) to loosen the two screws on the motor coupling. Do not remove these screws entirely; just loosen them by one or two turns of the hex driver. Usually it is necessary to hold the motor coupling in place with one hand while loosening the screws with the other hand.
, provided) to loosen the two screws on the motor coupling. Do not remove these screws entirely; just loosen them by one or two turns of the hex driver. Usually it is necessary to hold the motor coupling in place with one hand while loosening the screws with the other hand.



 .
.



 into the test solution.
into the test solution.
 .
.






 (
( ).
).






 ) in either the clockwise or counter-clockwise direction.
) in either the clockwise or counter-clockwise direction. .
. of the display value selected using the rotation rate control knob. It is normal for the last one or two digits on the display to flicker.
of the display value selected using the rotation rate control knob. It is normal for the last one or two digits on the display to flicker.


 ratio, which is the ratio compatible with Pine potentiostats. Other ratios are available for use with other potentiostats (see
ratio, which is the ratio compatible with Pine potentiostats. Other ratios are available for use with other potentiostats (see  is applied across the MOTOR STOP banana jacks. If the two banana jacks are shorted together (i.e., if the MOTOR STOP stop signal is driven to ground), then the motor stops rotating.
is applied across the MOTOR STOP banana jacks. If the two banana jacks are shorted together (i.e., if the MOTOR STOP stop signal is driven to ground), then the motor stops rotating.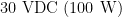 and the other is
and the other is 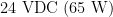 . These power supplies are located within the high voltage section of the control unit (see below).
. These power supplies are located within the high voltage section of the control unit (see below).

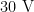 or
or 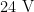 ) is to be replaced (see above).
) is to be replaced (see above).