


Highly Sensitive Electrochemical Determination of Lead in Tap Water
Last Updated: 1/10/23 by Alex Peroff
Download as PDF1Abstract
Wang, J.; Tian, B. Screen-printed stripping voltammetric/potentiometric electrodes for decentralized testing of trace lead. Anal. Chem., 1992, 64(15), 1706–1709.
Florence, T. M. Anodic stripping voltammetry with a glassy carbon electrode mercury-plated in situ. J. Electroanal. Chem. Interfacial Electrochem., 1970, 27(2), 273-281. Inexpensive and disposable screen-printed carbon electrodes are first coated with mercury microdroplets (microns in diameter) by reduction of mercury acetate (Hg2+) at a constant electrode potential. Then, lead ions (Pb2+) in a sample are reduced and collected into the mercury droplets. This pre-concentration step allows determination of very low concentrations (parts per billion, ppb) of lead when anodic differential pulse voltammetry (DPV)
2Introduction
3Background
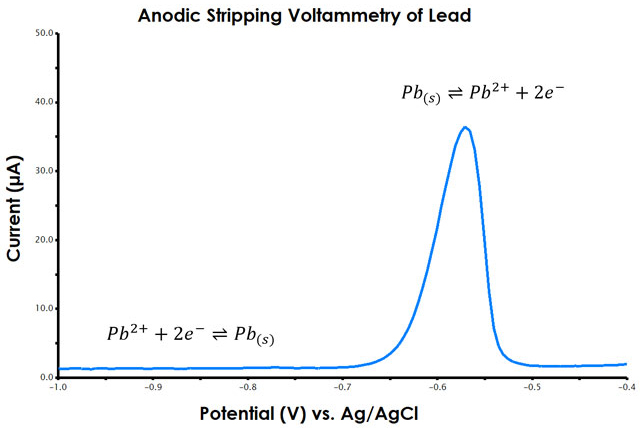
Figure 1. Anodic Stripping Voltammetry (ASV) of Lead
 |
(1) |

Figure 2. Bare Carbon Screen-Printed Electrode and Mercury-Plated Screen-Printed Electrode
 |
(2) |
4Differential Pulse Voltammetry
5Solution Preparation
- Nitric Acid Solution (3.0 M) - Prepare 1 L of 3.0 M nitric acid solution. For this solution, you will need concentrated nitric acid (HNO3, 68.9%). CAUTION: Nitric acid is highly corrosive. Avoid contact with skin. If any contacts the skin, rinse profusely with copious amounts of water. Into a 1 L volumetric flask and under constant stirring, slowly add 190 mL of concentrated HNO3 to approximately 700 mL of water. Dilute to the mark with distilled water. Label the solution as "3 M HNO3 Solution". The nitric acid solution is for cleaning all glassware.
- Hydrochloric Acid Solution (1.0 M) - Prepare 100 mL of 1.0 M hydrochloric acid solution. For this solution, you will need high purity, concentrated hydrochloric acid (HCl, 37%). CAUTION: Hydrochloric acid is highly corrosive. Avoid contact with skin. If any contacts the skin, rinse profusely with copious amounts of water. Add 8.3 mL of concentrated HCl to approximately 70 mL of deionized water. Mix gently and dilute to the mark with deionized water. Label this solution as "1 M HCl Solution".
- Mercury Acetate (4 mM) in 0.2 M Hydrochloric Acid Solution - Prepare 100 mL of 4.0 mM mercury acetate (Hg(CH3COO)2, MW = 318.7 g/mol) in 0.2 M HCl acid solution. In a 100 mL volumetric flask, add 20 mL of 1 M HCl solution to approximately 50 mL deionized water. Dissolve 0.13 g Hg(CH3COO)2 into the solution. Dilute to the mark with deionized water. Label this solution as "4 mM Hg(CH3COO)2 in 0.2 M HCl Solution".
- Mercury Acetate (0.4 mM) in 20 mM Hydrochloric Acid Solution - Prepare 100 mL of 0.4 mM Hg(CH3COO)2 in 20 mM acid solution by dilution of the 4 mM Hg(CH3COO)2 in 0.2 M HCl solution. In a 100 mL volumetric flask, add 10 mL of the 4 mM Hg(CH3COO)2 in 0.2 M HCl solution and dilute to the mark with deionized water. Label this solution as "0.4 mM Hg(CH3COO)2 in 0.2 mM HCl Solution".
- Sodium Acetate (NaOAc) Buffer (1.0 M) - Prepare 100 mL of 1.0 M sodium acetate Buffer (Na(CH3COO), MW = 82.03 g/mol). Sodium acetate is abbreviated NaOAc. In a 100 mL volumetric flask, dissolve 8.20 g NaOAc in 50 mL deionized water. Add 4.2 mL of concentrated HCl and dilute to the mark with deionized water. Label this solution as "1.0 M NaOAc buffer".
- Sodium Acetate (NaOAc) Buffer (20 mM) - Prepare 1 L of 20 mM sodium acetate buffer. Prepare this solution by dilution of the 1.0 M NaOAc buffer previously prepared. In a 1 L volumetric flask, add 20 mL of the 1.0 M NaOAc buffer to approximately 700 mL deionized water and dilute to the mark. Label this solution as "20 mM NaOAc Buffer".
- 1000 ppm Lead Acetate (1000 ppm) in 20 mM NaOAc Buffer - Prepare 100 mL of 1000 ppm (4.83 mM) lead acetate (Pb(CH3COO)2, MW = 325.29 g/mol) in 20 mM NaOAc buffer. In a 100 mL volumetric flask, dissolve 0.18 g of Pb(CH3COO)2 in 70 mL of 20 mM NaOAc buffer, then dilute to the mark with 20 mM NaOAc buffer. Label this solution as "1000 ppm Pb2+in 20 mM NaOAc Buffer".
- Lead Acetate (1 ppm) in 20 mM NaOAc Buffer - Prepare 100 mL of 1 ppm (4.83 μM) in 20 mM NaOAc buffer by dilution of the 1000 ppm Pb(CH3COO)2 buffer solution previously prepared. In a 100 mL volumetric flask, add 100 μL of the 1000 ppm Pb2+ in NaOAc buffer to ~70 mL of the 20 mM NaOAc buffer. Mix gently then dilute to the mark with 20 mM NaOAc buffer. Label this solution "1 ppm Pb2+ in 20 mM NaOAc Buffer".
6Setup and Preparation
6.1Physical Setup
 WaveNowXV Potentiostat Bundles
compact voltammetry kit,
WaveNowXV Potentiostat Bundles
compact voltammetry kit,
 Screen-Printed Electrode Cell Starter Kit
large volume compact voltammetry cell (AKSPEJAR),
Screen-Printed Electrode Cell Starter Kit
large volume compact voltammetry cell (AKSPEJAR),
 Screen-Printed Electrode Large Volume Cell
and 5x4 mm carbon screen-printed electrodes.
Screen-Printed Electrode Large Volume Cell
and 5x4 mm carbon screen-printed electrodes.
 Carbon Screen-Printed Electrodes
Carbon Screen-Printed Electrodes
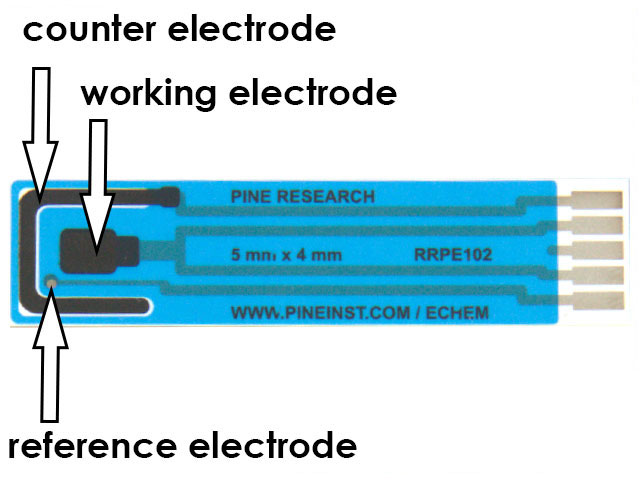
Figure 3. 5x4 mm Carbon Screen-Printed Electrode
6.2Mercury Deposition

Figure 4. Carbon Screen-Printed Electrode Connected to Grip Mount

Figure 5. Cell setup for mercury deposition onto carbon electrode

Figure 6. All Components for this Laboratory, Including Compact Voltammetry Cell, Potentiostat and Stirrer
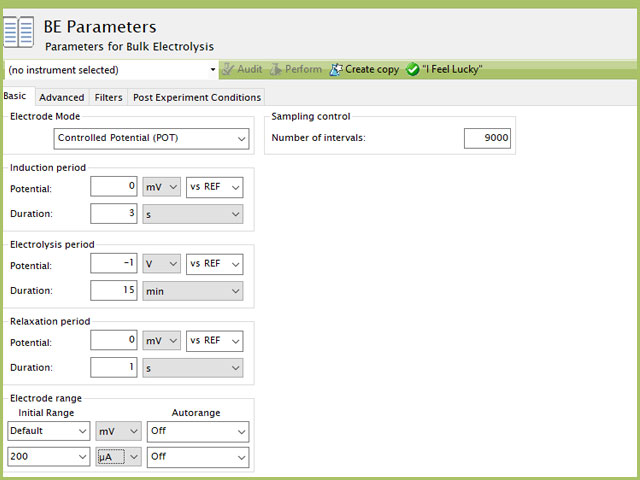
Figure 7. Bulk Electrolysis Parameters for Mercury Deposition in AfterMath
6.3General DPSV Analysis Protocol
- Add a stir bar and 50 mL of test solution to the large volume compact voltammetry electrochemical cell. Insert the screen-printed electrode (plated with Hg) into the hand grip (see Figure 4) and assemble the hand grip and cap into the cell.
- Degas the solution by sparging with N2 for at least 10 min. Turn the three way valve to flow N2 through the purge line. Continuously bubble N2 through the solution while constantly stirring at a fixed speed (about 500 RPM). Do not change the stir bar rate for future steps in the experiment. An easy way to accomplish this is to keep the stirring rate knob fixed in position and simply turn the stirrer off when not in use.
- After the solution has been purged for 10 min, the user will complete an anodic stripping voltammetry with DPV experiment (DPSV) (see Figure 8 for suggested DPSV parameters).
- The first step in DPSV is to concentrate lead ion into the mercury film (deposition period in AfterMath, also called pre-concentration). During this step, maintain both stirring and N2 bubbling. Monitor the experimental progress in AfterMath. After the deposition period (300 s, as shown in Figure 8), the DPV stripping waveform will start immediately. Turn off the stirrer and adjust the three way purge valve so N2 enters the cell through the blanket line. The gas from the blanket line should fill the cell head-space, not agitate solution. When you discontinue stirring, turn the power off on the stirrer. DO NOT adjust the RPM knob.
- After the experiment has completed, rename or reorganize the DPSV experiment accordingly in AfterMath to indicate the contents of the sample (e.g., 0 ppm Pb2+, pure water).
- After measuring the initial sample solution, add an aliquot of 100 μL 1 ppm Pb2+(equivalent to 2 ppb increment in Pb2+ concentration; see Table 1) to the electrochemical cell and repeat steps 4 and 5. You do not need to repeat the initial purging of solution for 10 minutes, as doing so is only necessary for the initial 50 mL of solution.
- Follow the analysis procedure for all solutions listed in Table 1. Data sufficient for calibration curve construction will be complete when the total added Pb2+ concentration is 10 ppb.
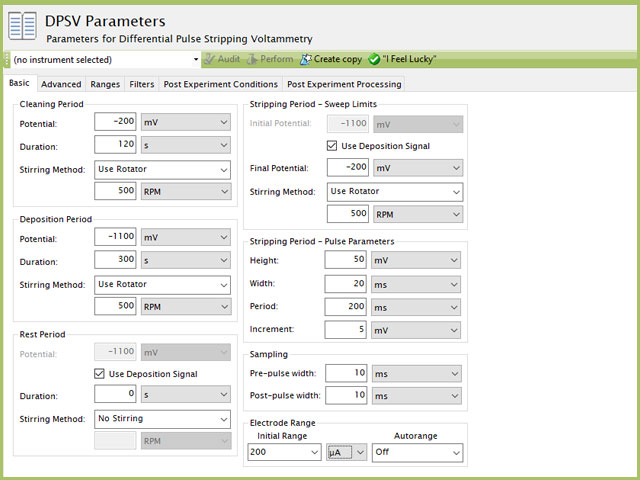
Figure 8. Differential Pulse Stripping Voltammetry (DPSV) Parameters in AfterMath
| Sample # | 1 ppm Pb2+ added to Buffer Standard (µL) | Total 1 ppm Pb2+ in Buffer Standard (µL) | Known [Pb2+] (ppb) |
| 1 | 0 | 0 | 0 |
| 2 | 100 | 100 | 2 |
| 3 | 100 | 200 | 4 |
| 4 | 100 | 300 | 6 |
| 5 | 100 | 400 | 8 |
| 6 | 100 | 500 | 10 |
6.4Standard Addition Method
7Experimental
7.1Measurement of Lead in Pure Water by Standard Addition
7.2Unknown Analysis
8Data Analysis
- Find the DPSV peak height (current) in AfterMath for each sample where Pb2+ was present (five samples for pure water, all six samples for the unknown). Record the peak current for each sample in a spreadsheet program.
- Prepare an overlay plot. In an overlay plot, all six DPSV voltammograms should be place into a single plot (see Figure 10).
- Perform a standard addition linear calibration analysis. Plot peak current vs. known concentration of Pb2+ as points (see Figure 11). Perform regression analysis on the data and draw a linear curve of best fit through the data points and record the linear equation of the line. A correlational coefficient > 0.98 is expected. Extrapolate the best fit line to the x-axis, where y = 0.
- From the slope of the regression, determine the Pb2+ concentration. This will correspond to the point on the extrapolated regression curve that crosses the x-axis. This is the concentration of Pb2+ in your unknown sample.
8.1Determine DPSV Peak Height
- In AfterMath, right-click the data trace that contains a peak of interest.
- Select "Add Tool", then select "Peak Height". Three colors or orbs will appear on the plot. The green orb should be moved onto the peak apex, the highest point in the peak. You can use the zoom (+ magnifying glass) in Aftermath to fine tune your selection, as well as the scroll wheel if using a mouse. The pink orbs are the baseline points and the black orbs extend the length of your baseline.
- Right-click the pink orb and select properties. From the menu, choose “free horizontal” as the method by which the baseline is drawn. Click OK. Drag and drop the pink orb (notice, a horizontal line is drawn through the pink orb) until the baseline is best drawn through the zero current leading and trailing baseline.
- The peak current will be displayed with a line drawn to the green dot.
- Record the peak height and known Pb2+ concentration in a spreadsheet program.
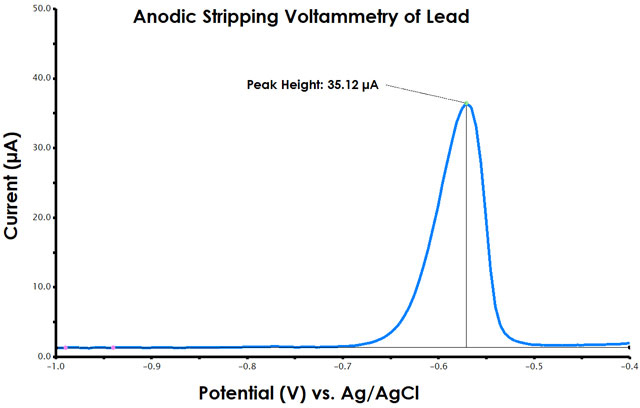
Figure 9. Anodic Stripping Voltammetry Peak Height Determination in AfterMath
8.2Prepare an Overlay Plot
- Right-click a node in the archive tree structure and select "New", then select "Plot".
- For each standard addition, including the first where no standard was added, click the “+” sign next to the DPSV Experiment icon. Click the “Voltammogram” icon.
- Click the data trace (the voltammogram) and copy it (either press Ctrl+C, or select Copy from the edit menu, or right-click the data and select copy).
- Click the new plot icon you created in step 1, then click the blank plot in the plot window. Paste the data (either press Ctrl+V, or select Paste from the edit menu, or right-click the data and select paste).
- Repeat steps 2 – 4 for all DPSV obtained in an experiment. Adjust axes as appropriate.
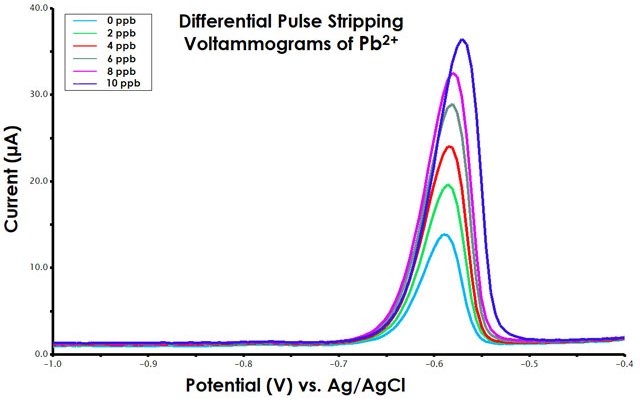
Figure 10. Differential Pulse Stripping Voltammograms for Standard Addition of Pb2+
8.3Construct Standard Addition Plot
- Plot peak current (from section 8.1) on the y-axis and known concentration of Pb2+ on the x-axis.
- Display the data as points, no connecting line.
- Perform regression analysis on the data points; i.e., add a trend line and view the resulting equation and correlational coefficient. Record the regression (linear) equation. The correlational coefficient should be > 0.98.
- For DPSV in pure water, the intercept will be close to (0,0) since one would not expect current when no Pb2+ was present to strip. For DPSV of unknown samples, the intercept will not be zero.
- Extrapolate the regression line to the x-axis (when y = 0 for the linear equation).
![Standard addition plot for [Pb2+] in tap water by differential pulse stripping voltammetry](https://www.pineresearch.com/shop/wp-content/uploads/sites/2/2019/04/Lead-lab-fig-11-2.jpg)
Figure 11. Standard Addition Plot for [Pb2+] in Tap Water by Differential Pulse Stripping Voltammetry
8.4Determine Unknown Lead Concentration
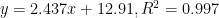 |
(3) |
9Questions
- What is the unknown concentration of lead in the water analyzed? If drinking water was analyzed, how does the concentration compare to the legal limit in public drinking water?
- What is the purpose of purging and blanketing with nitrogen gas? What might you expect would happen if this step was not performed?
- Why is the stir bar used in the pre-concentration step of a differential pulse anodic stripping voltammetry experiment? What do you expect would be the result if the solution was not stirred during this step?
- Write the electrochemical half reaction of interest that occurs during the mercury plating process, the pre-conditioning step, and the stripping step of DPSV.
- Stripping voltammetry can detect several metals at the same time, as long as their thermodynamic electrode potentials are separated by about 100 mV. Draw the stripping voltammogram you would expect for a mixture of Cd2+, Pb2+, and Cu2+ (HINT: look up the appropriate half reactions in a textbook).
10References
- Bard, A. J.; Faulkner, L. A. Electrochemical Methods: Fundamentals and Applications, 2nd ed. Wiley-Interscience: New York, 2000.
- Wang, J.; Tian, B. Screen-printed stripping voltammetric/potentiometric electrodes for decentralized testing of trace lead. Anal. Chem., 1992, 64(15), 1706–1709.
- Florence, T. M. Anodic stripping voltammetry with a glassy carbon electrode mercury-plated in situ. J. Electroanal. Chem. Interfacial Electrochem., 1970, 27(2), 273-281.